



Ang RT-qPCR ay binuo mula sa ordinaryong teknolohiya ng PCR.Nagdaragdag ito ng mga fluorescent na kemikal (fluorescent dyes o fluorescent probes) sa tradisyunal na sistema ng reaksyon ng PCR, at nakikita ang proseso ng PCR annealing at extension sa real time ayon sa kanilang iba't ibang mekanismo ng luminescent.Ang mga pagbabago sa fluorescent signal sa medium ay ginagamit upang kalkulahin ang dami ng pagbabago ng produkto sa bawat cycle ng PCR.Sa kasalukuyan, ang pinakakaraniwang pamamaraan ay fluorescent dye method at probe method.
Paraan ng fluorescent dye:
Ang ilang mga fluorescent dyes, tulad ng SYBR Green Ⅰ, PicoGreen, BEBO, atbp., ay hindi naglalabas ng liwanag nang mag-isa, ngunit naglalabas ng fluorescence pagkatapos mag-binding sa minor groove ng dsDNA.Samakatuwid, sa simula ng reaksyon ng PCR, hindi matukoy ng makina ang fluorescent signal.Kapag ang reaksyon ay nagpapatuloy sa annealing-extension (two-step method) o extension stage (three-step method), ang double strands ay bubuksan sa oras na ito, at ang bagong DNA polymerase Sa panahon ng strand synthesis, ang mga fluorescent molecule ay pinagsama sa dsDNA minor groove at naglalabas ng fluorescence.Habang tumataas ang bilang ng mga cycle ng PCR, parami nang parami ang mga dyes na pinagsama sa dsDNA, at ang signal ng fluorescent ay patuloy ding pinapahusay.Kunin ang SYBR Green Ⅰ bilang isang halimbawa.
Paraan ng pagsisiyasat:
Ang Taqman probe ay ang pinakakaraniwang ginagamit na hydrolysis probe.Mayroong fluorescent group sa 5′ dulo ng probe, kadalasang FAM.Ang probe mismo ay isang sequence na pantulong sa target na gene.Mayroong fluorescent quenching group sa 3′ dulo ng fluorophore.Ayon sa prinsipyo ng fluorescence resonance energy transfer (Förster resonance energy transfer, FRET), kapag ang reporter fluorescent group (donor fluorescent molecule) at ang quenching fluorescent group (acceptor fluorescent molecule) Kapag ang excitation spectrum ay nagsasapawan at ang distansya ay napakalapit (7-10nm), ang autoscitasi ng donor fluorescent molecule ay maaaring magdulot ng mahinang pagtanggap ng fluorescent molecule. natapos.Samakatuwid, sa simula ng reaksyon ng PCR, kapag ang probe ay libre at buo sa system, ang reporter fluorescent group ay hindi maglalabas ng fluorescence.Kapag ang pagsusubo, ang panimulang aklat at probe ay nagbubuklod sa template.Sa yugto ng extension, ang polymerase ay patuloy na nag-synthesize ng mga bagong chain.Ang DNA polymerase ay may 5′-3′ exonuclease na aktibidad.Kapag naabot ang probe, i-hydrolyze ng DNA polymerase ang probe mula sa template, ihihiwalay ang reporter fluorescent group mula sa quencher fluorescent group, at ilalabas ang fluorescent signal.Dahil may one-to-one na relasyon sa pagitan ng probe at template, ang probe method ay mas mataas kaysa sa dye method sa mga tuntunin ng katumpakan at sensitivity ng pagsubok.
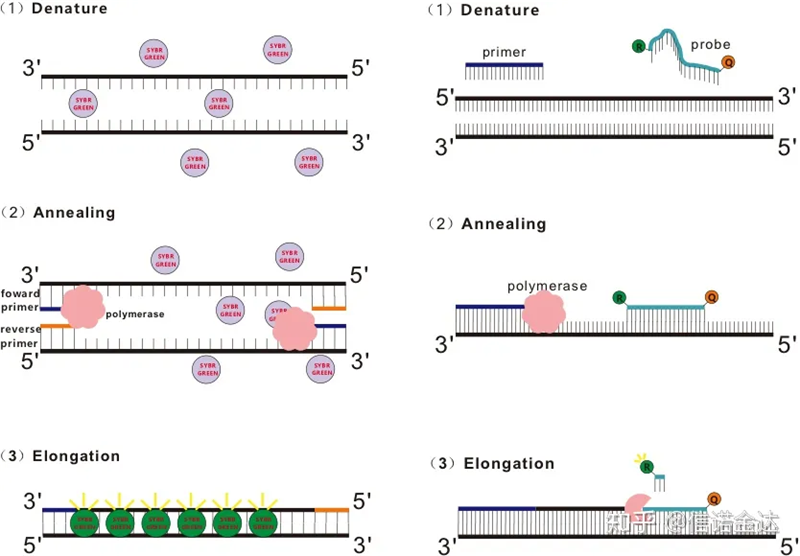
Fig 1 Prinsipyo ng qRT-PCR
Pangunahing disenyo
Mga Prinsipyo:
Ang mga panimulang aklat ay dapat na idinisenyo sa konserbadong rehiyon ng serye ng nucleic acid at may pagtitiyak.
Pinakamainam na gumamit ng cDNA sequence, at ang mRNA sequence ay tinatanggap din.Kung hindi, alamin ang disenyo ng cds region ng DNA sequence.
Ang haba ng fluorescent quantitative na produkto ay 80-150bp, ang pinakamahaba ay 300bp, ang haba ng primer ay karaniwang nasa pagitan ng 17-25 base, at ang pagkakaiba sa pagitan ng upstream at downstream na mga primer ay hindi dapat masyadong malaki.
Ang nilalaman ng G+C ay nasa pagitan ng 40% at 60%, at 45-55% ang pinakamaganda.
Ang halaga ng TM ay nasa pagitan ng 58-62 degrees.
Subukang iwasan ang mga primer na dimer at self-dimer, (huwag lumabas ng higit sa 4 na pares ng magkasunod na complementary base) hairpin structure, kung hindi maiiwasan, gawin ang ΔG<4.5kJ/mol* Kung hindi mo matiyak na ang gDNA ay naalis sa panahon ng reverse transcription Malinis, pinakamahusay na idisenyo ang mga primer ng intron *3′ na dulo upang maiwasan ang mga istrukturang AT, 3′ na tuluy-tuloy, at mayaman sa rehiyon ng A/GC (T/G/G) ) mga panimulang aklat at hindi
tiyak Ang homology ng heterogeneously amplified sequence ay mas mainam na mas mababa sa 70% o may 8 complementary base homology.
Database:
CottonFGD paghahanap sa pamamagitan ng mga keyword
Primer na disenyo:
IDT-qPCR primer na disenyo
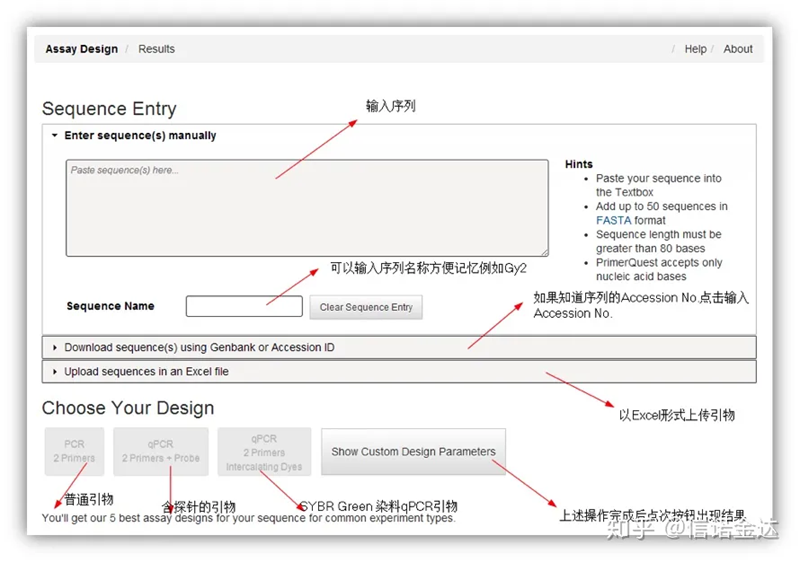
Fig2 IDT online primer design tool page
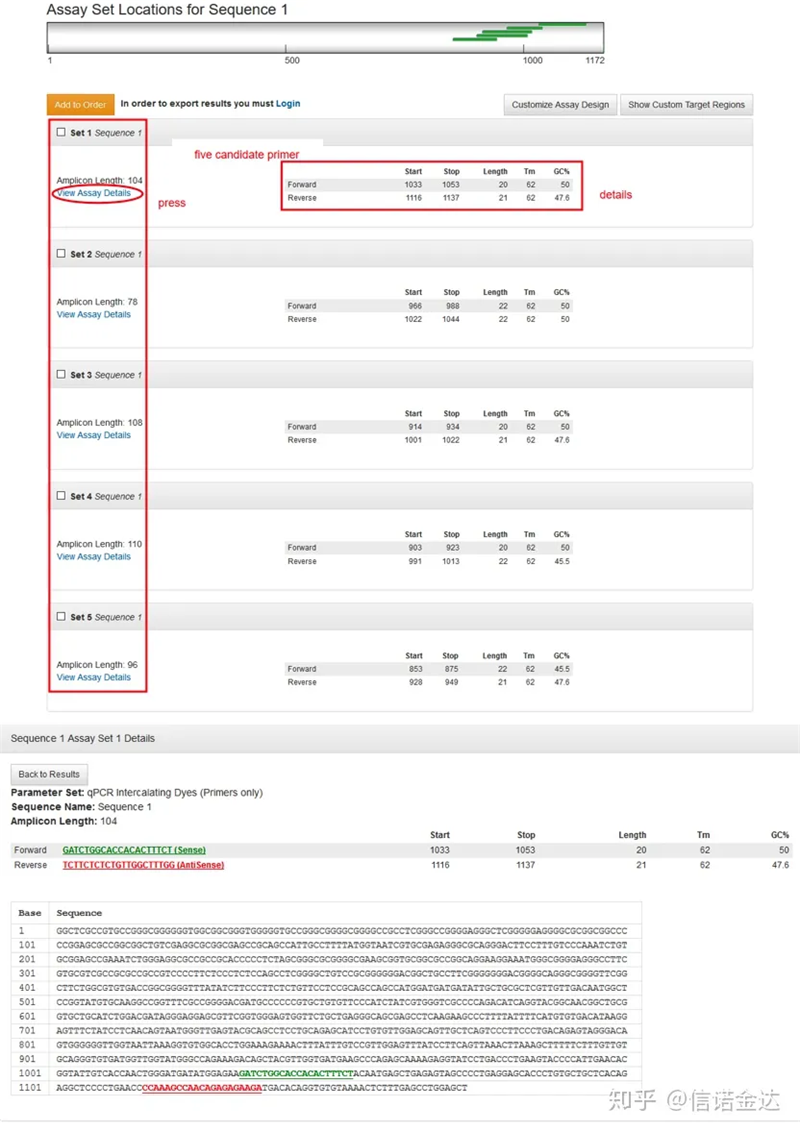
Pagpapakita ng pahina ng resulta ng Fig3
Disenyo ng lncRNA primers:
lncRNA:ang parehong mga hakbang bilang mRNA.
miRNA:Ang prinsipyo ng paraan ng stem-loop: Dahil ang lahat ng miRNA ay maiikling sequence na humigit-kumulang 23 nt, hindi maisagawa ang direktang PCR detection, kaya ginagamit ang stem-loop sequence tool.Ang stem-loop sequence ay isang single-stranded DNA na humigit-kumulang 50 nt, na maaaring bumuo ng isang hairpin structure nang mag-isa.3 'Ang dulo ay maaaring idisenyo bilang isang sequence na pantulong sa miRNA na bahagyang fragment, pagkatapos ay ang target na miRNA ay maaaring konektado sa stem-loop sequence sa panahon ng reverse transcription, at ang kabuuang haba ay maaaring umabot sa 70bp, na naaayon sa haba ng amplified na produkto na tinutukoy ng qPCR.Tailing miRNA primer na disenyo .
Pagtukoy na partikular sa amplification:
Online blast database: CottonFGD blast ayon sa pagkakapareho ng pagkakasunod-sunod
Lokal na sabog: Sumangguni sa paggamit ng Blast+ para gawin ang local blast, ang linux at macos ay maaaring direktang magtatag ng lokal na database, ang win10 system ay maaari ding gawin pagkatapos mag-install ng ubuntu bash.Lumikha ng database ng lokal na sabog at sa lokal na sabog;buksan ang ubuntu bash sa win10 .
Pansinin: Ang upland cotton at sea island cotton ay mga tetraploid na pananim, kaya ang resulta ng pagsabog ay kadalasang dalawa o higit pang mga posporo.Noong nakaraan, ang paggamit ng mga NAU cd bilang isang database upang magsagawa ng pagsabog ay malamang na makahanap ng dalawang homologous na gene na may kaunting pagkakaiba sa SNP.Karaniwan, ang dalawang homologous na gene ay hindi maaaring paghiwalayin ng panimulang disenyo, kaya ang mga ito ay itinuturing na pareho.Kung mayroong isang halatang indel, ang panimulang aklat ay karaniwang idinisenyo sa indel, ngunit ito ay maaaring humantong sa pangalawang istraktura ng panimulang aklat Ang libreng enerhiya ay nagiging mas mataas, na humahantong sa pagbaba sa kahusayan ng amplification, ngunit ito ay hindi maiiwasan.
Pagtuklas ng panimulang pangalawang istraktura:
Mga hakbang:open oligo 7 → input template sequence → isara ang sub-window → save → hanapin ang primer sa template, pindutin ang ctrl+D para itakda ang haba ng primer → pag-aralan ang iba't ibang pangalawang istruktura, tulad ng self-dimerization body, heterodimer, hairpin, mismatch, atbp. Ang huling dalawang larawan sa Figure 4 ay ang mga resulta ng pagsubok ng mga primer.Ang resulta ng front primer ay mabuti, walang halatang dimer at hairpin na istraktura, walang tuluy-tuloy na komplementaryong base, at ang ganap na halaga ng libreng enerhiya ay mas mababa sa 4.5, habang ang back primer ay nagpapakita ng tuluy-tuloy Ang 6 na base ay pantulong, at ang libreng enerhiya ay 8.8;bilang karagdagan, lumilitaw ang isang mas seryosong dimer sa dulong 3′, at lumilitaw ang isang dimer ng 4 na magkakasunod na base.Kahit na ang libreng enerhiya ay hindi mataas, ang 3′ dimer Chl ay maaaring seryosong makaapekto sa pagtitiyak ng amplification at kahusayan ng amplification.Bilang karagdagan, ito ay kinakailangan upang suriin para sa mga hairpins, heterodimer, at mismatches.
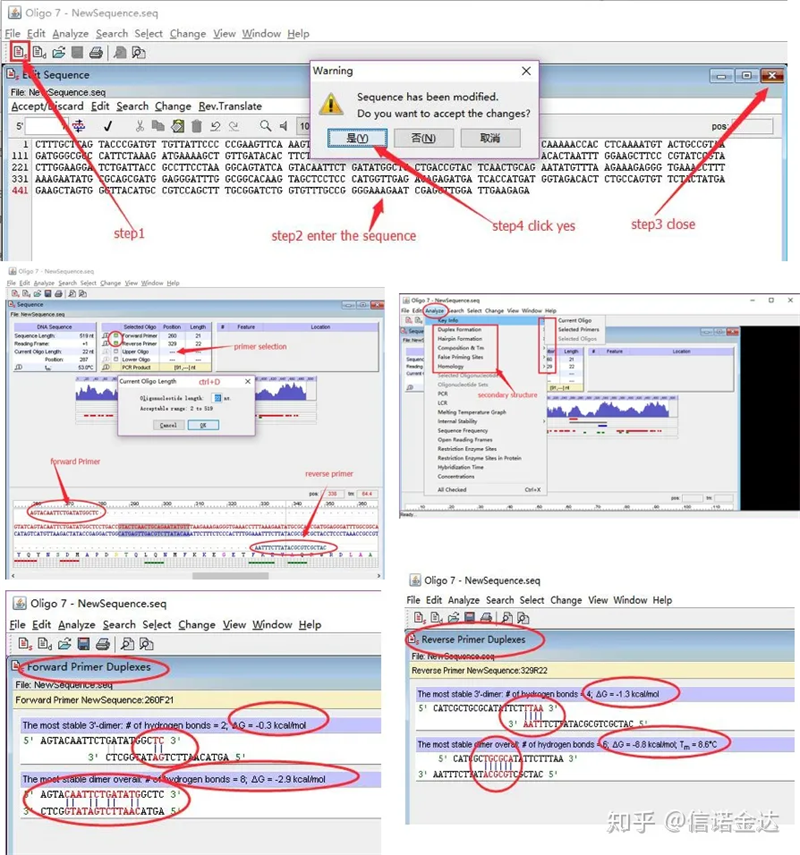
Mga resulta ng pagtuklas ng Fig3 oligo7
Pagtukoy sa kahusayan ng amplification:
Ang kahusayan ng amplification ng reaksyon ng PCR ay seryosong nakakaapekto sa mga resulta ng PCR.Gayundin sa qRT-PCR, ang kahusayan ng amplification ay partikular na mahalaga para sa dami ng mga resulta.Alisin ang iba pang substance, machine at protocol sa reaction buffer.Ang kalidad ng mga panimulang aklat ay mayroon ding malaking impluwensya sa kahusayan ng amplification ng qRT-PCR.Upang matiyak ang katumpakan ng mga resulta, ang parehong relatibong fluorescence quantification at ang absolute fluorescence quantification ay kailangang makita ang amplification efficiency ng mga primer.Kinikilala na Ang epektibong qRT-PCR amplification efficiency ay nasa pagitan ng 85% at 115%.Mayroong dalawang mga pamamaraan:
1. Karaniwang paraan ng curve:
a.Paghaluin ang cDNA
b.gradient dilution
c.qPCR
d.Linear regression equation para kalkulahin ang kahusayan ng amplification
2. LinRegPCR
Ang LinRegPCR ay isang programa para sa pagsusuri ng real time na RT-PCR Data, na tinatawag ding quantitative PCR (qPCR) data batay sa SYBR Green o katulad na chemistry.Gumagamit ang program ng hindi baseline na data na naitama, nagsasagawa ng baseline correction sa bawat sample nang Hiwalay, tinutukoy ang isang window-of-linearity at pagkatapos ay gumagamit ng linear regression analysis upang magkasya sa isang tuwid na linya sa PCR data set.Mula sa slope ng linyang ito ay kinakalkula ang kahusayan ng PCR ng bawat indibidwal na sample.Ang ibig sabihin ng kahusayan ng PCR sa bawat amplicon at ang halaga ng Ct bawat sample ay ginagamit upang kalkulahin ang panimulang konsentrasyon sa bawat sample, na ipinahayag sa mga arbitrary na fluorescence unit.Ang input at output ng data ay sa pamamagitan ng Excel spreadsheet.Sample lang
kailangan ang paghahalo, walang gradient
kailangan ang mga hakbang:(Kunin ang Bole CFX96 bilang isang halimbawa, hindi masyadong Machine na may malinaw na ABI)
eksperimento:ito ay isang karaniwang eksperimento sa qPCR.
output ng data ng qPCR:Makikilala ng LinRegPCR ang dalawang anyo ng mga output file: RDML o quantification Amplification result.Sa katunayan, ito ay ang real-time na halaga ng pagtuklas ng numero ng cycle at fluorescence signal ng makina, at ang amplification ay nakuha sa pamamagitan ng pagsusuri sa fluorescence change value ng linear segment na kahusayan.
Pagpili ng data: Sa teorya, ang halaga ng RDML ay dapat na magagamit.Tinatantya na ang problema ng aking computer ay hindi makilala ng software ang RDML, kaya mayroon akong excel output value bilang orihinal na data.Inirerekomenda na magsagawa muna ng magaspang na screening ng data, tulad ng pagkabigo sa pagdaragdag ng mga sample, atbp. Ang mga puntos ay maaaring tanggalin sa output data (siyempre, hindi mo maaaring tanggalin ang mga ito, hindi papansinin ng LinRegPCR ang mga puntong ito sa susunod na yugto)
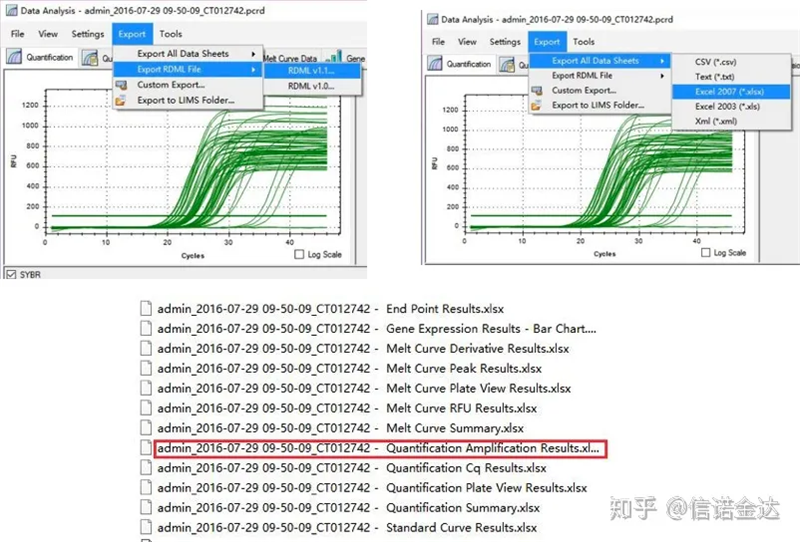
Fig5 qPCR na pag-export ng data
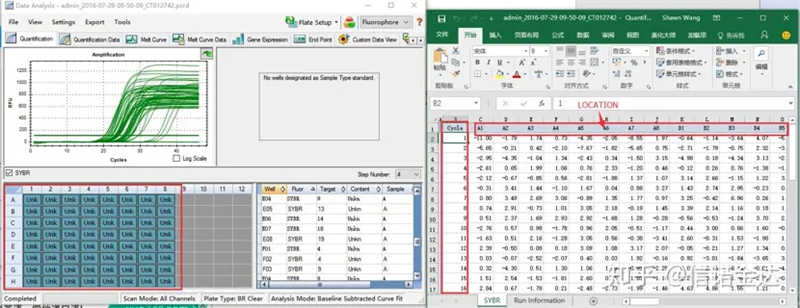
Fig6 pagpili ng mga sample ng kandidato
Pag lagay ng datos:Buksan ang mga resulta ng amplification ng kwalipikasyon.xls, → buksan ang LinRegPCR → file → basahin mula sa excel → piliin ang mga parameter tulad ng ipinapakita sa Figure 7 → OK → i-click ang tukuyin ang mga baseline
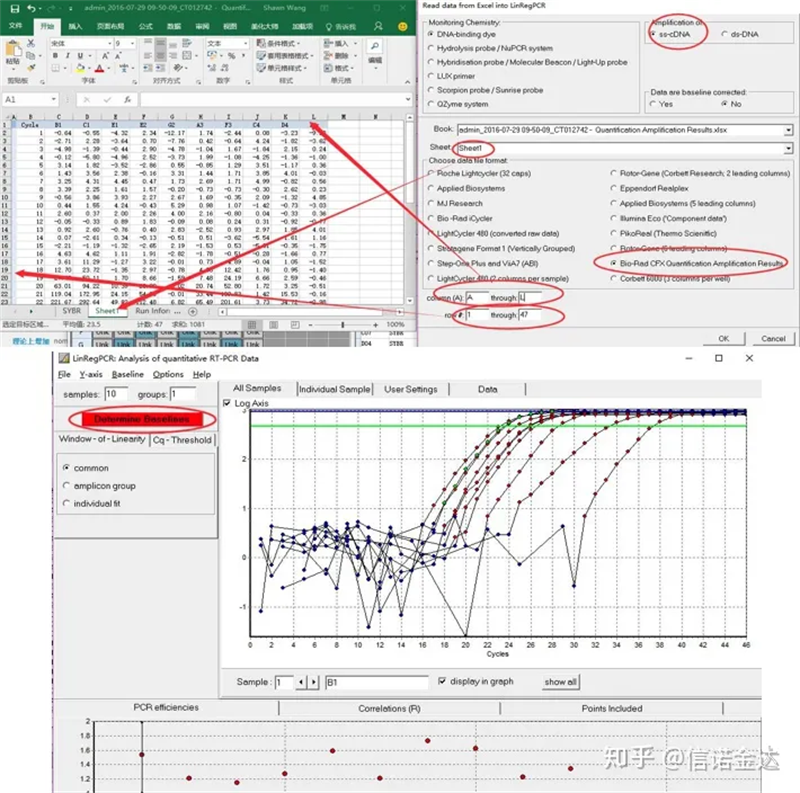
Fig7 hakbang ng linRegPCR data input
Resulta:Kung walang pag-uulit, hindi kailangan ang pagpapangkat.Kung mayroong pag-uulit, ang pagpapangkat ay maaaring i-edit sa sample na pagpapangkat, at ang pangalan ng gene ay ipinasok sa identifier, at pagkatapos ay ang parehong gene ay awtomatikong ipangkat.Panghuli, mag-click sa file, i-export ang excel, at tingnan ang mga resulta.Ang kahusayan sa amplification at R2 na mga resulta ng bawat balon ay ipapakita.Pangalawa, kung hahatiin mo sa mga grupo, ang naitama na average na kahusayan sa amplification ay ipapakita.Tiyakin na ang kahusayan ng amplification ng bawat primer ay nasa pagitan ng 85% at 115%.Kung ito ay masyadong malaki o masyadong maliit, nangangahulugan ito na ang kahusayan ng amplification ng panimulang aklat ay mahina.
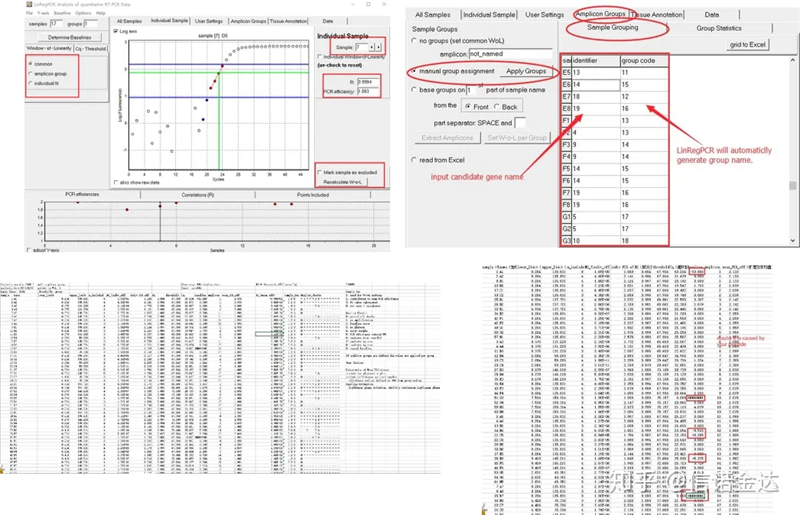
Fig 8 Resulta at output ng data
Eksperimental na proseso:
Mga kinakailangan sa kalidad ng RNA:
kadalisayan:1.72.0 ay nagpapahiwatig na maaaring mayroong natitirang isothiocyanate.Ang malinis na nucleic acid A260/A230 ay dapat nasa paligid ng 2 . Kung may malakas na pagsipsip sa 230 nm, ito ay nagpapahiwatig na mayroong mga organikong compound tulad ng phenate ions.Bilang karagdagan, maaari itong makita ng 1.5% agarose gel electrophoresis.Ituro ang marker, dahil ang ssRNA ay walang denaturation at ang molecular weight logarithm ay walang linear na relasyon, at ang molecular weight ay hindi maipahayag nang tama.Konsentrasyon: Sa teoryahindimas mababa sa 100ng/ul, kung ang konsentrasyon ay masyadong mababa, ang kadalisayan ay karaniwang mababa at hindi matangkad
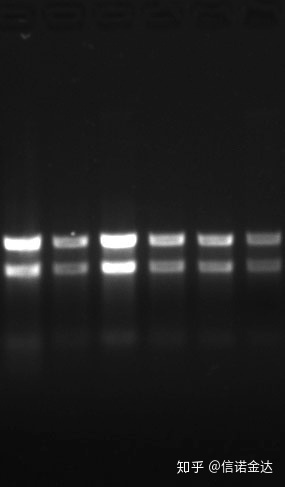
Fig9 RNA gel
Bilang karagdagan, kung ang sample ay mahalaga at ang konsentrasyon ng RNA ay mataas, inirerekumenda na aliquot ito pagkatapos ng pagkuha, at palabnawin ang RNA sa isang pangwakas na konsentrasyon na 100-300ng/ul para sa reverse transcription.Saang proseso ng reverse transcription, kapag na-transcribe ang mRNA, ang mga primer na oligo (dt) na maaaring partikular na magbigkis sa mga polyA tails ay ginagamit para sa reverse transcription, habang ang lncRNA at circRNA ay gumagamit ng random na hexamer (Random 6 mer) primer para sa reverse transcription ng kabuuang RNA Para sa miRNA, ang miRNA-specific na neck-loop primer ay ginagamit para sa reverse transcription.Maraming kumpanya ang naglunsad na ngayon ng mga espesyal na tailing kit.Para sa stem-loop method, ang tailing method ay mas maginhawa, high-throughput, at reagent-saving, ngunit Ang epekto ng pagkilala sa mga miRNA ng parehong pamilya ay hindi dapat kasing ganda ng stem-loop method.Ang bawat reverse transcription kit ay may mga kinakailangan para sa konsentrasyon ng gene-specific primers (stem-loops).Ang panloob na sanggunian na ginamit para sa miRNA ay U6.Sa proseso ng stem-loop inversion, ang isang tubo ng U6 ay dapat na baligtarin nang hiwalay, at ang harap at likod na mga primer ng U6 ay dapat na direktang idagdag.Ang parehong circRNA at lncRNA ay maaaring gumamit ng mga HKG bilang panloob na sanggunian.Sapagtuklas ng cDNA,
kung walang problema sa RNA, dapat maayos din ang cDNA.Gayunpaman, kung gagawin ang pagiging perpekto ng eksperimento, pinakamahusay na gumamit ng panloob na reference gene (Reference gene, RG) na maaaring makilala ang gDNA mula sa mga cd.Sa pangkalahatan, ang RG ay isang housekeeping gene., HKG) tulad ng ipinapakita sa Figure 10;Noong panahong iyon, gumagawa ako ng soybean storage protein, at gumamit ng actin7 na naglalaman ng mga intron bilang panloob na sanggunian.Ang laki ng amplified fragment ng primer na ito sa gDNA ay 452bp, at kung cDNA ang ginamit bilang template, ito ay 142bp.Pagkatapos ay natuklasan ng mga resulta ng pagsubok na ang Bahagi ng cDNA ay talagang nahawahan ng gDNA, at pinatunayan din nito na walang problema sa resulta ng reverse transcription, at maaari itong magamit bilang isang template para sa PCR.Walang silbi na magpatakbo ng agarose gel electrophoresis nang direkta sa cDNA, at ito ay isang nagkakalat na banda, na hindi nakakumbinsi.
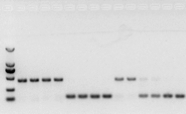
Fig 10 pagtuklas ng cDNA
Ang pagpapasiya ng mga kondisyon ng qPCRsa pangkalahatan ay walang problema ayon sa protocol ng kit, higit sa lahat sa hakbang ng halaga ng tm.Kung ang ilang mga panimulang aklat ay hindi mahusay na idinisenyo sa panahon ng disenyo ng panimulang aklat, na nagreresulta sa isang malaking pagkakaiba sa pagitan ng halaga ng tm at ng teoretikal na 60°C, inirerekomenda na ang cDNA Pagkatapos ng mga sample ay halo-halong, magpatakbo ng isang gradient PCR na may mga primer, at subukang iwasan ang pagtatakda ng temperatura nang walang mga banda bilang ang halaga ng TM.
Pagsusuri sa datos
Ang conventional relative fluorescence quantitative PCR processing method ay karaniwang ayon sa 2-ΔΔCT.Template ng pagproseso ng data.
Kaugnay na Mga Produkto:
Real Time PCR EasyTM –SYBR GREEN I
RT Easy I (Master Premix para sa unang strand cDNA synthesis)
RT Easy II(Master Premix para sa unang strand cDNA synthesis para sa qPCR)
Oras ng post: Mar-14-2023



